

View the January 2022 Case of the Month below:
Clinical Cases from Emory Division of Hospital Medicine Annual Clinical Vignette Conference
“A muscle weakness with dysphagia”
Submitted by:
Anup Patel, MD
Assistant Professor and Senior Physician
Division of Hospital Medicine
Emory Department of Medicine
Edited by Yelena Burklin, MD, FHM, FACP
CASE
A 40-year-old female with a history of Wolf Parkinson White syndrome presented with a subacute progressive muscle weakness in the proximal upper and lower extremities bilaterally that was associated with ulcerative skin lesions on the chest and thighs, as well as a solid food dysphagia. Her vital signs upon admission to the hospital were within normal limits, and a focused physical examination revealed patchy hypopigmented areas in various stages of scarring that were located on the anterior and posterior aspects of the upper trunk, along with the erythematous active ulcerative lesions in the axillary areas bilaterally. She was noted to have bilateral hand synovitis in multiple joints and a proximal 3/5 upper and lower extremity muscle weakness bilaterally.
Laboratory findings were notable for creatine kinase (CK) 4848 U/L, AST 200 IU/L, ALT 75 IU/L, ESR 45 mm/hr, CRP 73 mg/L, +ANA 1:1280 Speckled, high positive anti-NXP-2 (nuclear matrix protein 2) antibody, and an elevation of the following: aldolase, anti-RNP, anti-Smith, and anti-SSA IgG antibodies.
Muscle biopsy was performed and the results are provided below:
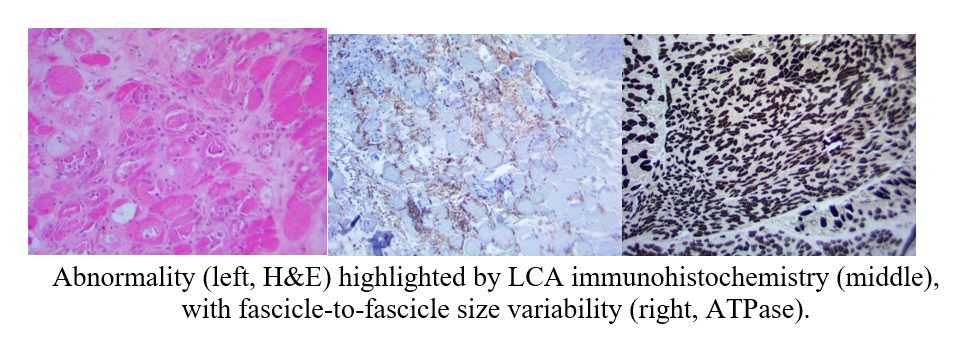
Pathology report was consistent with a presence of degenerating and atrophic fibers, myo-phagocytosis, myocyte necrosis and no regeneration or primary inflammation.
The regional increase in connective tissue suggested the presence of a process for months to years.
What is your diagnosis?
FINAL DIAGNOSIS
Necrotizing Autoimmune Myopathy (NAM), also known as the Immune Mediated Necrotizing Myopathy (IMNM).
Hospitalization course:
The patient has been treated with a pulse dose of IV steroids, Mycophenolate Mofetil, and Intravenous Immunoglobulin (IVIG) therapy. Alternative treatment options such as methotrexate, tacrolimus, cyclosporine, or cyclophosphamide were not applied at that time.
Upon completion of the initial treatment, there was an observed improvement in CK level from over 4000 U/L to 350 U/L, and inflammatory markers had normalized within two months following hospital presentation.
During a follow-up physical examination, the patient continued to have limited ability to raise both arms to a shoulder level, but she could stand up from a wheelchair with additional support and ambulate several steps with assistance.
The patient remained dependent upon steroids and IVIG therapy, and given late presentation, she might have suffered a long-term disability with persistent weakness despite intense immunosuppressive treatment.
DISCUSSION
Necrotizing Autoimmune Myopathy (NAM), also known as the Immune-Mediated Necrotizing Myopathy (IMNM), is a rare autoimmune-mediated inflammatory necrotizing disorder with the prevalence of approximately 9–14 cases per 100,000 people. It often carries a worse prognosis when it affects people at a younger age.
The disease is characterized by the presence of acute or subacute severe, symmetrical, proximal muscle weakness, usually associated with muscle-specific antibodies (anti-HMGCR or anti-SRP). The condition typically presents with persistently elevated CK levels, usually in the range of several thousand.
In contrast to the inflammatory myopathies, NAM or IMNM muscle biopsies often reveal no or minimal inflammation but prominent muscle fiber necrosis and regeneration.
Proximal muscle weakness, which is usually more prominent in the lower limbs, can be associated with neck muscle weakness and dysphagia. Respiratory muscle weakness is common and can result in hypercapnic respiratory failure and the need for mechanical ventilation. This manifestation can be observed not only in the setting of generalized muscle weakness but also at disease onset and serve as the only presenting symptom.
Two main autoantibodies associated with NAM or IMNM, are the anti-HMGCR (3-hydroxy-3-methylglutaryl-coenzyme A reductase) and anti-SRP (anti-signal recognition particle) antibodies, but the majority of patients with the disease have no detectable HMGCR or SRP auto-antibodies.
The prognosis in NAM/IMNM is worse than in the other types of myositis or myopathy due to the extent of muscle necrosis.
Initial differential diagnosis frequently includes dermatomyositis or lupus. NAM/IMNM presentation can mimic autoimmune dermatomyositis, and laboratory findings can be suggestive of lupus/dermatomyositis overlap syndrome.
Even though NAM/IMNM can resemble other autoimmune myopathies, the extent of muscle necrosis and a paucity of inflammatory infiltrate seen on muscle biopsy, distinguish it from other autoimmune myopathies.
This scenario highlights the importance of timely diagnosis of the immune-mediated necrotizing myopathy because it is often associated with an underlying malignancy. Therefore, cancer screening is required upon diagnosis. Proper identification of an underlying malignancy and early initiation of oncologic therapy offers life-prolonging measures earlier in the course of the disease. This type of myopathy should be considered in any patient presenting with an unexplained bilateral proximal muscle weakness, dysphagia, or respiratory compromise.
CITATIONS
Be the first to comment on "Faculty Development Case of the Month: January 2022"